QTL Café Tutorial
INTRODUCTION
Welcome to the QTL Café Tutorial. By the time you reach this page, you will have selected one of four examples and the Java applet will have appeared in front of you. What next? How do you use this example and how do you load your own data, analyse them and interpret the results?
The QTL café is designed to be easy to use and to provide informative analyses of data from pure line crosses such as doubled haploid lines, recombinant inbred lines, F2s and backcrosses. An example of each of these population types is included for you to use to explore the facility before you load your own data.
|
Example 1 - |
Brassica - doubled haploid lines (DHLs) of Brassica oleracea |
|
Example 2 - |
Arabidopsis - recombinant inbred lines (RILs) from Arabidopsis thaliana |
|
Example 3 - |
Simulated F2 data |
|
Example 4 - |
Simulated Backcross data |
This tutorial will illustrate the analysis of Example 1, but the processes involved apply to all of the examples. At the end are details of how to prepare your own data for loading into the software, with a checklist of the options which you may need to use and a list of 'error' messages which can appear during loading.
If you wish to analyse Example 1 whilst referring to this tutorial on screen but you have already selected another example, you will need to reload the applet. Go back, using the browser, to the page listing the examples and choose Example 1. The applet will reload with the correct data set. When this tutorial page is reopened the applet window will be hidden behind it but it should be accessible from the status bar.
The tutorial was written whilst using the software with Netscape Navigator 4.5, on a PC with Windows 95 so those of you using different systems may find that details differ slightly. If you find differences that other users of your system would find useful to know, please contact us.
THE QTL ANALYSIS APPLET
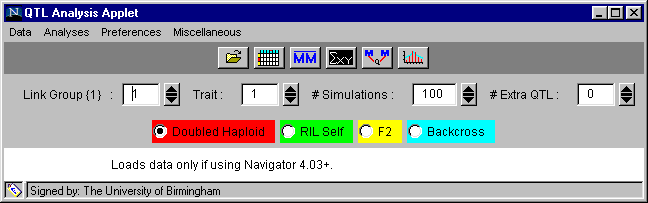
We will first give you an overview of the options available on the applet and then go into more detail as we work through the example. The applet window will always appear as shown in Figure 1.
Figure 1. The applet window set for example 1, Brassica DHLs, linkage group 1 and trait 1.
Beneath the title bar are five horizontal fields controlling different aspects of analysis.
|
Data |
Analyses |
Preferences |
Miscellaneous |
|
Load |
Marker Means |
Means as Symbols |
Trait Distribution |
|
Edit |
Marker Regression |
Kosambi Mapping Function |
Map ® |
|
Sort Markers |
Interval Mapping |
File Format |
Draw |
|
|
Expected |
Calculate Trait |
Add QTL |
|
|
|
Allow Dragging |
Clear List |
|
|
|
Colours |
About |
Clicking on one of the choices in a menu will activate it and cause the menu to disappear. Three of the options in the Preferences menu (Means as Symbols, Kosambi Mapping Function and Allow Dragging) are 'checked' items - if you select one by clicking on it, it will be activated and the menu will disappear but if you look at the menu again this item will appear with a tick beside it showing that it is functioning. Clicking on the item again will de-select it and the tick will be removed.
|
Open folder |
= load data |
|
Spreadsheet |
= view and edit data |
|
MM |
= single marker ANOVA |
|
S XY |
= marker regression analysis |
|
M-Q-M |
= interval mapping analysis |
|
Bar chart |
= expected |
|
Link group { } |
choose the linkage group you wish to examine |
|
Trait |
choose the trait you wish to examine |
|
Simulations |
choose the number of simulations you wish to use to provide confidence intervals for the marker regression analysis |
|
Extra QTL |
choose the number of additional QTL above the default single QTL you wish to fit to the given linkage group (i.e. the value 0 here means that 1 QTL will be fitted.) |
Field 4: 4 radio buttons, one for each of the four population types:
Doubled haploid RILSelf F2 Backcross
The default is doubled haploid, but you must remember to select the correct one for the population you are analysing. If you look at any of the other examples, you must specify the population type with these buttons before doing the analyses.
USING THE APPLET TO WORK THROUGH THE AN EXAMPLE
Example 1 involves 150 DHLs of Brassica oleracea, 2 quantitative traits and 84 markers across the 9 linkage groups. These data for the line means for each trait will be automatically loaded for you when you start. We will explain how to load your own data later.
Prior analysis
Before the data were loaded, an ANOVA was performed with a standard statistical package to establish that the lines differed significantly for these quantitative traits and to obtain a value for the additive genetical variance, VA.
Viewing and editing data
The data for this example are already loaded and can be viewed in spreadsheet form using either the 'edit' facility or the spreadsheet button. You can move around the spreadsheets using the scroll bars.
The marker data are in the larger spreadsheet and the trait data are in the smaller one. Clicking on a spreadsheet will bring it to the front. A spreadsheet can be moved around the screen by clicking and dragging on the title bar. Each row has the data for a different line.
Marker sheet:
|
Row 1 |
Value. This box enables you to change genotype data - see below. |
|
Row 2 |
Marker name. Although marker names of any length are acceptable, the whole name may not be visible due to the cell size. |
|
Row 3 |
Position of marker in centimorgans (cM) on the linkage group (chromosome) shown on Row 3. |
|
Row 4 |
Linkage group on which the marker is positioned |
|
Row 5 |
Number of lines with missing genotype data for that marker. This helps in the decision of whether to exclude a marker from the analyses. |
|
Row 6 |
Segregation ratio of the two alleles, given as nAA:nBB. This will indicate if there is segregation distortion for that marker. Where there are no BBs, e.g. Example 4, backcross data, the ratio will appear as nAA:0. |
|
Row 7 etc |
Genotype data. In this example, the data are in the form: AA = 1, BB = 3, Missing = 4. There are no heterozygotes (AB = 2) as the lines are doubled haploids. Other notations are acceptable (see loading data) and the data will appear in the notation that you entered. |
|
Column 1 |
The line (or individual) number, which is assigned in the order in which the data are entered. |
Trait sheet:
|
Row 1 |
Trait names, if these have been entered. The size of the cell will prevent viewing of the whole of a long name. |
|
Row 2 etc. |
Trait means. Missing values are in the form '-' but other notations are acceptable (see loading data). |
|
Column 1 |
The line (or individual) number, which is assigned in the order in which the data is entered. |
|
Column 2 etc. |
Each trait appears in a different column, in the order in which they were loaded. |
Markers and/or lines (individuals) can be selected for exclusion from the analyses. Use the scroll bars on the spreadsheet to bring the marker name or line number into view. Move the cursor to the marker name in Row 2 or the line number in the first column and double-click. The cell will turn red, signifying that this marker or line has been excluded. Double-clicking on a red cell will deselect it, returning it to normal colour and including the marker or line in subsequent analyses.
Reasons for selecting markers: Although the genetic map of B. oleracea that we used had 310 loci, the data for only 84 of the loci were entered.
Changing genotype values: Genotype scores can be changed within the spreadsheet.
Displaying and saving results
Once an analysis has been selected, the results are displayed in a window, similar to the one shown in Fig 2, which is from the analysis of linkage group 2 with trait 1 by Marker Regression.
N.B. Do cancel a window when not needed as it takes up RAM. Since the windows can be hidden behind the browser window, it is a good idea to minimise the browser so that the Applet can be used like a toolbar and output can be seen at all times. In Windows 95 all current results windows are shown on the status bar.
Windows can be moved around the screen by clicking and holding the left hand mouse button on their blue title bar and dragging the window to the required position. This enables you to put windows side by side to compare their results.
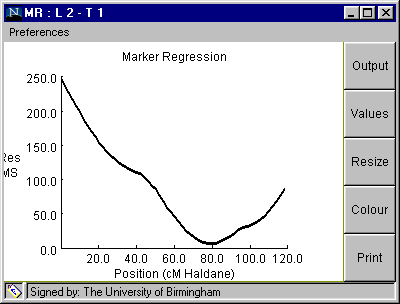
Fig 2. Typical analysis output window.
At the top right of the window are the usual minimise, maximise and cancel buttons. At the top left is a title, with an abbreviation of the analysis, the linkage group and the trait if applicable. On the right hand side are five buttons and under the title bar is a 'preferences' menu. These five buttons allow the following operations.
|
Output |
toggles between graph and text output. Maximise the window and/or use the scroll bars to see the full content of the text output. |
|
Values |
tabulates x and y co-ordinates on the text output area |
|
Resize |
resizes the window to its original shape if it has been shrunk, stretched or maximised. Shrinking or stretching can be achieved by moving the mouse arrow to the edge of the window. When it changes to a double headed arrow, click and hold down the left hand mouse button and drag the window edge to the required position. |
|
Colour |
toggles between the default colours of the graph and black and white. Alternative colours can be selected using the 'colour' option in the 'preferences' menu on the Applet itself. |
|
|
sends the contents of the window directly to the printer. The size of the printout of the graph depends on the size of the window. The size of text output does not change with window size. The title bar and the buttons do not appear on the printout. |
|
Axes |
clicking on 'preferences' on the menu bar below the title bar of the window will reveal an 'axes' option which allows you to change the axes settings. Clicking on 'axes' will show a table giving the maximum, minimum and interval points on the x and y axes. Clicking in a cell will allow you access to alter its value but you must press the return or enter key for the change to be recognised. When you have made all your changes click in the 'apply' box, to produce a tick, and then cancel the axes window. The axes will change to your settings once the axes window has been cancelled. |
A Java security window may appear when you try to view the text output or if you print directly from a window. It will warn that the applet is requesting permission for the high risk activity of reading and writing to the system clipboard for your computer or for the low risk activity of printing. Click on 'grant'.
The text output is written directly to your clipboard. You can paste the contents of the clipboard straight into another application such as a word processing package. A result window containing a graph can be copied to the clipboard by pressing Alt and Print Screen on the keyboard simultaneously and it can then be pasted into a document, but in this case the whole window complete with title bar and buttons will be copied. If several windows are open, only the currently selected one, with the blue title bar, will be copied. It is useful to have a 'Word' document open, or minimised on the status bar, to receive each result that you wish to save. When the text is pasted it may not appear as on screen and you may need to insert or remove tab spaces or change the font size to align the headings and figures correctly.
The display in the 'marker means' and 'expected' results windows can be changed from vertical bars to square symbols by selecting Means as Symbols in the Preferences menu, if desired. If selected, this item will appear with a tick, when the menu is next viewed.
Prior to analysis
Operators are: + - / * ^ (to the power of, i.e. 2^3 = 8), functions are LN (natural logarithm) LOG (base 10 logarithm) SQRT (square root) EXP (exponential) and constants are PI (3.1415927) and E (2.7182818). Round brackets can be used to group calculations. The precedence of operators is that normally found in computing languages so that multiplication/division has a higher precedence over adding and subtracting e.g. 1+2*3 = 7 and (1+2)*3 = 9
When in doubt, use brackets. The letter T followed by a number n indicates that the value of the nth trait should be substituted in the calculation, thus T2*PI will multiply trait two by PI (3.1415927). The enter key must be pressed for the entry to be registered and the 'Apply' box must be checked for the equation to be used in subsequent analyses. You must uncheck the Apply box before you can work with the next trait.
An example of how to enter a complex expression into the trait calculator is:
PI*SQRT(EXP(LN(T1))^2) press enter, click apply then OK. T1 indicates trait 1, squaring cancels the square root and the exponential function cancels the natural logarithm so the result is trait 1 multiplied by PI. Obviously, T1*PI will achieve the same outcome but this illustrates how you can build up an expression.
QTL Analysis
You are now in a position to analyse the data for the trait and linkage group you have selected. For this example we have selected, linkage group (i.e. chromosome) 2 with trait 1 because this linkage group has a QTL for trait 1.
Single marker ANOVA
Using the 'marker means' option in the Analyses menu or the MM button, a preliminary analysis can be carried out. This looks at all individual associations between markers and phenotype.
Theory: For each marker, the mean of the trait scores for all those lines homozygous for the allele from parent 1 is compared to the mean trait score for all those lines homozygous for the allele from parent 2 and to the mean of those heterozygous for this marker.
Results of analysis
When the analysis has been selected, a result window will appear (Fig 3), with a graph showing the additive effect associated with each of the nine markers in linkage group 2.
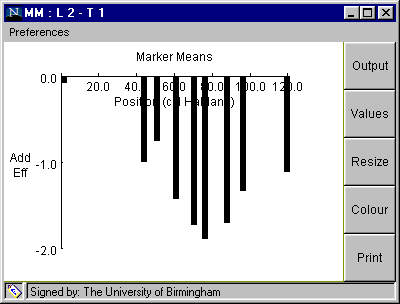
Fig. 3. Window with graphical output from "Marker Means" analysis
Interpretation of Window in Fig 3.:
Table 1. Data displayed on clicking "Output" button shown in Fig. 3.
Marker Positions and Trait Means:
Linkage Group: 2 Trait: 1 No. Pos. Add. eff. mean(11) n(11) mean(12) n(12) mean(22) n(22) F P 1 0.0 -0.08 32.29 74 0.0 0 32.45 54 0.018 0.8925 NS 2 42.4 -1.0 31.45 66 0.0 0 33.46 77 3.344 0.0696 NS 3 49.5 -0.76 31.67 65 0.0 0 33.19 83 1.843 0.1767 NS 4 59.5 -1.43 30.65 53 0.0 0 33.51 79 6.286 0.0134 * 5 68.8 -1.73 30.21 49 0.0 0 33.67 99 9.051 0.0031 ** 6 74.8 -1.9 30.09 52 0.0 0 33.9 95 11.303 0.0010 *** 7 86.5 -1.71 30.05 48 0.0 0 33.47 97 9.102 0.0030 ** 8 95.1 -1.34 30.66 42 0.0 0 33.33 104 4.72 0.0315 * 9 118.4 -1.12 31.17 50 0.0 0 33.41 91 3.52 0.0627 NS
Table 2 illustrates the corresponding form of the data for an F2 example in which heterozygote and dominance effects are dispalyed.
Table 2. As for Table 1 but for an F2 population.
Marker Positions and Trait Means Linkage Group: 1 Trait: 1 No. Pos. Add. eff. Dom. eff. mean(11) n(11) mean(12) n(12) mean(22) n(22) F P 1 0.0 0.6 -1.19 43.23 31 41.44 51 42.03 18 2.442 0.0923 NS 2 11.0 0.73 -1.2 43.4 28 41.46 52 41.93 20 2.75 0.0689 NS 3 22.0 1.55 -1.06 43.94 32 41.33 54 40.84 14 7.007 0.0014 ** 4 33.0 2.7 0.02 44.33 36 41.65 45 38.94 19 20.182 0.0 *** 5 44.0 3.86 0.8 44.8 36 41.75 48 37.08 16 52.919 0.0 *** 6 55.0 5.31 1.45 45.46 34 41.6 54 34.84 12 193.281 0.0 *** 7 66.0 3.65 0.27 45.13 29 41.75 55 37.82 16 38.048 0.0 *** 8 77.0 2.98 0.52 44.49 31 42.04 49 38.54 20 24.416 0.0 *** 9 88.0 3.21 0.84 44.66 26 42.3 54 38.24 20 27.84 0.0 *** 10 99.0 2.39 0.68 44.12 25 42.41 51 39.33 24 14.137 0.0 ***
Marker Regression

Fig. 4. The Marker Regression window
Table 3. Marker regression analysis from clicking on "OUTPUT" button on Fig. 4 (100 simulations).
Linkage Group: 2 Trait: 1 QTL located at 80.0 cM Additive effect = -1.9991219 Source df MS F P Add Reg 1 2355.68 75.22 0.0 Residual 7 6.62 0.21 0.838 Error 139 31.32 Simulated QTL position is 78.747 +/- 11.167 Simulated Additive QTL effect is -2.007 +/- 0.666
etc
Table 4. Marker regression analysis from clicking on "OUTPUT" button on Fig. 4 (1000 simulations).
QTL Mapping by Marker Regression: Result Linkage Group: 2 Trait: 1 QTL located at 80.0 cM Additive effect = -1.9991219 Source df MS F P Add Reg 1 2355.68 75.22 0.0050 Residual 7 6.62 0.21 0.834 Error 139 31.32 Simulated QTL position is 77.98 +/- 15.292 Simulated Additive QTL effect is -2.052 +/- 0.609
To plot this QTL on a map.
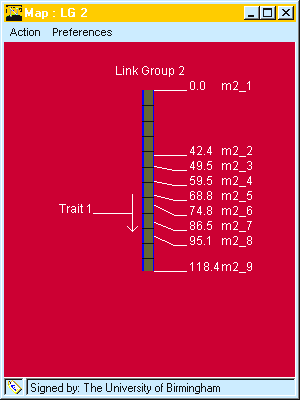
Fig. 5. QTL drawn to map. The direction of the arrow indicates it P1 has the decreasing allele, the horizontal line indicates the QTL location and the length of the arrow gives the confidence interval. Marker names and positions are also indicated.are .
Interval Mapping
This can be addressed by clicking the MQM button and results in the window shown in Fig 6.
The analysis follows the method described by Haley and Knott (1992).
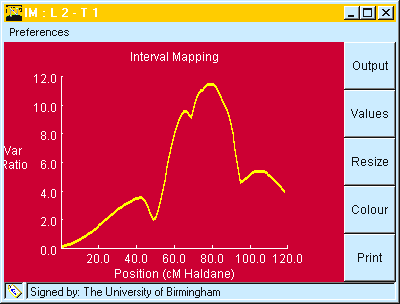
Fig. 6. Output from "Interval Mapping" showing Variance Ratio for the regression plotted against map. position.
Table 5. Output from clicking "OUTPUT" button on window shown in Fig 6.
QTL Mapping by Interval Mapping: Result 1 QTL
Linkage Group: 2
Trait: 1
QTL located at 79 cM
Test Statistics : F 11.493 LR 11.2146
Res. SS - Full Model 6262.181 d.f. 146
Res. SS - Red. Model 6755.136 d.f. 147
Mean 31.8945
Add Eff. -2.0071
Taken together ( Fig. 6 and Table 5) we find:
These values can be compared with those given above for Marker Regression in Table 2.
QTL can be added to the map as before.
Comparison of observed and expected marker means.
When either the Marker Regression or Interval Mapping analyses have been run, the observed marker means can be compared to their expected values (based on the mapped QTL and their additive effects) by use of the "Bar chart" button on the applet or by choosing "expected" from the analysis menu, as shown in Fig. 7.
The "Expected" graph will always be produced using the QTL locations and effects estimated in the last analysis attempted.
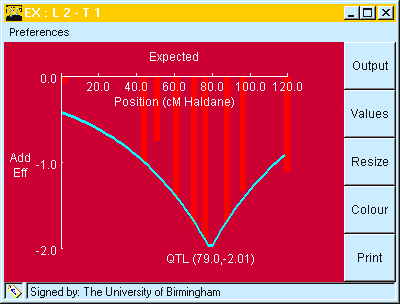
Fig. 7. Window showing observed marker means (Vertical bars) at different chromosome positions compared to their expected values (blue line). The expected values were taken (automatically) from the interval mapping analysis, shown in Table 5.
Candidate loci comparisons: If the "Allow dragging" item in the "preferences menu is checked, then it is possible to move the expected line by "click and drag" with the mouse to the position of the possible candidate locus. This allows you to see how well it fits the candidate.
Loading data
Once the Applet has appeared, after selecting any of the four example sets, your own data can be loaded.
Data files
Three data files are required; all with the same file name but with different extensions. At present, Unix users are advised to use lowercase letters for their directory and file names when using this software.
|
filename.gen - |
genotype data |
|
filename.mrk - |
marker data |
|
filename.tra - |
trait data |
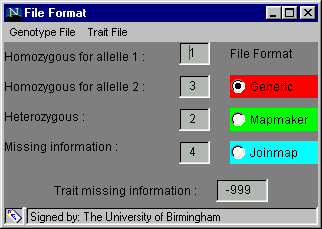
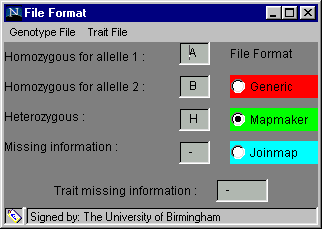
The software is designed to accept several different formats in order to minimise the number of alterations you will need to make to your data. The File Format dialog box, found in the Preferences menu and shown below, is used to inform the software of the format of your data, before you try to load it.
Options available under the headings on grey menu bar:
|
Genotype file |
Trait file |
|
- individuals as cols |
- traits as rows |
|
- marker names |
- trait names |
One of three formats, generic, Mapmaker or Joinmap, can be chosen by clicking on the radio button beside the name (the default option is generic). When one is selected, the default genotype coding and missing trait coding are inserted into the text boxes but these can be altered if your data has different codes. The default codings for the generic format and the Mapmaker format are shown above. To alter a code: click on the relevant cell, delete the entry, insert your own code, press enter or return for the change to be registered and move on to the next cell.
Genotype file
A generic genotype file is shown on the left below. The first row always contains three numbers, representing the number of individuals (n-ind), the number of markers (n_mrk) and the number of traits (n_tra). Then follows n_ind rows of individuals (lines), each row containing the genotype at each marker locus of the individual. The genotypes are coded: 1, homozygous for parent 1; 2, heterozygous; 3, homozygous for parent 2 and 4, missing data. The data can be delimited by tab characters, spaces or commas. Other codes will be accepted if you change the default code in the File Format box as explained above.
If your data has the individuals as columns (= markers as rows), click on 'individuals as cols' in the 'genotype file' menu in the File Format box, before loading the data.
If you have included marker names (at the top of each column in the default orientation or at the start of each row in the 'individuals as columns' option), click on 'marker names' in the 'genotype file' menu of the File Format box.
If you have not included marker names make sure the order of the markers is the same in the genotype file and the marker file. If marker names are included but the order is not the same in the two files, a message informing you that there are marker name mismatches will appear when you load the data. It will suggest that you use the menu option to sort the data. Clicking on OK at the end of the message does not sort the data, you must go to the 'data' menu on the Applet menu bar and select sort. The data will be loaded whether or not you select this option. Only use the sort option if you are sure the markers are in a different order in the two files. If you get the message when you think the order should be the same, check your data files carefully. The marker order in the marker file will be taken as correct and the .gen data sorted accordingly.
An example of a genotype file in Mapmaker format is shown below. An extra heading line giving the data type is included but you must define the population type in the Applet as well. Marker names are expected, prefaced with an * , and the genotype data for any individual are in a column. The options 'individuals as cols' and 'marker names' do not have to be selected.
A genotype file in Joinmap, Mapmaker and generic formats are shown below:
JOINMAP FORMAT
name = Arab popt = SSD nloc = 67 nind = 98 ntra = 3 # more comments # and again # next line 2 spaces #next line 2 tabs g4715-a abbaababbb abaab-abbb aa-bbbaaab baabbaabaa babbaabb-a babbbbbaaa baaaaababa bbbbaaaaaa bba-abaaaa bbabbbbb m488 abbaababbb abaab-abbb aaabbbaaab baabbaabaa babbaabb-a babbbbbaaa baaaaababa bbbbaaaaaa bbaaabaaaa bbabbbbb g3786 aaaaababab ababbaabbb aaabbbaaab baabbaa-aa babbaabb-a babbbaaaa& baaabababb bbabaaabba -ba-bbaaa- bbabbbbb g3829 aaaaababab ababbbabab aa-bb-aabb bbabbaaaab babbabab-a aaaabbaaaa baaa--babb bbbbaaabba -ba-bbb--a bbab--ab
etc.
MAPMAKER FORMAT
data type ri self 98 67 3 # more comments # and again # next line 2 spaces #next line 2 tabs *g4715-a abbaababbb abaab-abbb aa-bbbaaab baabbaabaa babbaabb-a babbbbbaaa baaaaababa bbbbaaaaaa bba-abaaaa bbabbbbb *m488 abbaababbb abaab-abbb aaabbbaaab baabbaabaa babbaabb-a babbbbbaaa baaaaababa bbbbaaaaaa bbaaabaaaa bbabbbbb *g3786 aaaaababab ababbaabbb aaabbbaaab baabbaa-aa babbaabb-a babbbaaaa baaabababb bbabaaabba -ba-bbaaa- bbabbbbb *g3829 aaaaababab ababbbabab aa-bb-aabb bbabbaaaab babbabab-a aaaabbaaaa
etc
GENERIC FORMAT
25 3 1 1, 3, 3 1, 3, 3 1, 1, 1 3, 3, 3 3, 3, 3 1, 3, 1 1, 1, 3 1, 1, 1 3, 3, 3 3, 3, 3 3, 3, 1 1, 1, 1 3, 3, 1 3, 3, 3 1, 1, 1 3, 1, 1 3, 3, 3 3, 3, 3 1, 1, 3 1, 1, 3 3, 3, 3 3, 3, 3 3, 3, 1 1, 1, 3 3, 3, 3
Marker File
The marker file contains all the markers you have chosen for all the linkage groups. Markers spaced approximately 10 to 15 cM apart are sufficient. Please read the section on page 4 concerning the choice of markers. Marker names can be any length but do not use spaces or commas within a name. If you are using data in Mapmaker format, do not include the asterisk (*) at the start of the marker name in the marker file.
Column 1 contains the marker names.
Column 2 contains the marker linkage groups denoted by an integer number starting at 1.
Column 3 contains the respective marker positions.
The data can be delimited by tab characters, spaces or commas.
A typical marker file is shown below for markers on 5 linkage groups.
g4715-a, 1, 0.0 m488, 1, 0.0 g3786, 1, 8.0 g3829, 1, 16.5 m235, 1, 20.1 m253, 1, 33.4 gapB, 1, 48.8 m213, 1, 63.2 g4026, 1, 72.5 m315, 1, 82.1 g4552, 1, 88.5 m532, 1, 103.4 g17311, 1, 109.2 m246, 2, 0.0 g4553, 2, 2.1 g4532, 2, 6.0 g4133, 2, 9.9 m216, 2, 16.0 m251, 2, 22.6 g6842, 2, 29.5 er, 2, 32.2 m220, 2, 38.0 m323, 2, 43.6 g17288, 2, 43.6 g4514, 2, 49.1 m336, 2, 52.9 m583, 3, 0.0 g4523, 3, 0.6 m228, 3, 7.6 g4708, 3, 7.6 m105, 3, 11.6 g4711, 3, 20.7 m249, 3, 37.8 g4117, 3, 37.8 g4564-b, 3, 41.2 g4014, 3, 43.9 m457, 3, 48.1 g2778, 3, 53.3 m424, 3, 57.3 g3843, 4, 0.0 g2616, 4, 4.9 m506, 4, 4.9 m518, 4, 14.9 pCITd23, 4, 18.1 g6837, 4, 26.6 g10086, 4, 27.7 g4564-a, 4, 27.7 m326, 4, 28.7 m226, 4, 32.0 g3845, 4, 32.5 m600, 4, 43.8 g8300, 4, 48.4 g3088, 4, 50.3 pCITd99, 4, 53.3 g3713, 4, 64.6 g3715, 5, 0.0 m217, 5, 4.5 g3837, 5, 5.5 CHS, 5, 10.7 g4560, 5, 17.1 m291, 5, 20.3 g4715-b, 5, 27.6 m247, 5, 58.1 g4028, 5, 62.5 m435, 5, 81.2 g2368, 5, 88.8 m555, 5, 94.7
Trait file.
The software expects a trait file with the following format: each column represents a single trait and contains the trait values for all the individuals; names of the individuals and trait names are not included.
If you include the trait name at the top of each column, click on 'trait names' in the 'trait file' menu in the File Format box before loading the data.
If you have the traits in rows rather than columns (with the trait name at the start of the row, if included), click on 'traits as rows' in the 'trait file' menu in the File Format box (and 'trait names', if included).
If you are using a code for missing trait values that is different to the default one in the option you are choosing in the File Format box, remember to alter the code in the box before loading the data. (Never use zero as a code for missing trait values).
The individuals in the trait file should be in the same order as in the genotype file.
The data can be delimited by tab characters, spaces or commas. A typical trait file for 3 traits (columns) is shown below:
248.1 238.75 263.6 348.4 357.2 268.6 371.7 353.2 336.7 317.7 351.75 282.4 367.7 370.3 336.2 430.7 423.7 362.7 276.7 281.2 240 211.1 240.5 194.33 449.3 445.8 345.6 248.8 226.3 247 407.7 441.3 351.8 230.2 171.7 184.2 195.9 171.3 187.2 362.3 390.8 318.3
etc
Check list
Error messages found when loading data:
Error: trait exception: Unconvertible string 'Mean-1' has occurred at line 1 of the trait file
The trait headings (the first of which was Mean-1) had been left in the trait file. Either remove the headings or go to Preferences then File format then trait file and click on trait names. Then reload the data.
Error: trait exception: Unconvertible string '-' has occurred at line 8 of the trait file
The missing data in this trait file should have been entered as '-999' as these files were in the generic format but one on line eight was entered as '-'. This data point could be corrected but if all the missing data were entered as '-', they could either be replaced with ' -999' or the missing trait value in the box in the generic file format could be changed from '-999' to '-' (via Preferences, then File format)
Error: trait exception: null occurred when the 'tab' between two trait entries in a trait file was deleted, effectively causing the loss of a data point.
Error: An error has occurred in the genotype file header line
This message occurred when loading data in Mapmaker format without having changed the File format found in Preferences to Mapmaker.
It also occurred when entering data in generic form without the header line in the genotype file which specifies how many individuals, markers and traits there are.
Error: Unexpected character '1064' has occurred at line 2 of the genotype file
The name of the individual 1064 had been left at the start of line 2 in the genotype file (generic format). Names of individuals should be deleted.
Error: Unexpected character '11' has occurred at line 4 of the genotype file
One of the data points in line 4 of a generic genotype file had been entered wrongly as 11 instead of 1. Any unexpected character in the genotype file will be notified in this way with the line number specified (the header line is included in the count). Similarly an entry of 'c' instead of 'a' in a Mapmaker format genotype file will generate a similar message.
Error: Unexpected character m1-1 has occurred at line 2 of the genotype file
It is possible to include marker names in a generic format genotype file if Marker names is selected via Preferences, File format, Genotype file. An error message with the first marker name identified will occur if this selection is not done.
If a mistake is made in the genotype header line such that the numbers of individuals, markers or traits specified do not correspond with the numbers of individuals, markers or traits in the data, error or information messages will be generated. For example:
When entering the Arabidopsis data, which has 98 individuals, 67 markers and 9 traits, in Mapmaker format the following messages were displayed:
Information: 67 marker name mismatches, use menu option to sort; obtained when the number of individuals was entered wrongly as 100 (but the sort option should not be used in this case).
Error: Trait exception: null; obtained when the number of individuals was wrongly entered as 97.
Error; but with no explanation, was obtained when the number of markers was wrongly entered as either 65 or 69.
Error: Trait exception: null; obtained when the number of traits was wrongly entered as 8 or 10.
When entering the Brassica data in generic format without marker names in the genotype file or trait names the following messages were obtained when errors were made in the genotype header line:
Error; but with no explanation, was obtained when the number of individuals was wrongly entered as 149 instead of 150 and also when the number of markers was entered as either 82 or 86 instead of 84.
Error: Trait exception: null; obtained when the number of traits was entered wrongly.
N.B. When the number of individuals was entered as 180 instead of 150, the data was accepted. When the data was viewed via the spreadsheet button on the applet, the data for these non-existent individuals were displayed as stars in the genotype boxes and 0.0 in the trait boxes.
The following mistakes created in a Mapmaker format genotype file resulted in:
Information: 66 marker name mismatches, use menu option to sort. (The number relates to the number of markers affected although only one or two may be wrong)
Error: [but no explanation] occurred when a data point was omitted from a generic genotype file.
Information: 1 marker name mismatches, use menu option to sort.
Because both the genotype and marker files in the Mapmaker format contain the marker names, a mistake in a marker name in either will be identified. If the markers with their data have been entered in a different order in the two files the sort option can be used but if the error is a typing error of the marker name just correct the mistake. When marker names are included in a generic genotype file similar error messages will be obtained if the marker names in the genotype and marker files do not agree.
Error: Unconvertible string '8.0' has occurred at line 3 of the marker file was obtained when a linkage group was omitted from the third entry in a Mapmaker or generic format marker file . A similar message is obtained if a marker position is omitted.
The most common error is a missing data point in the genotype file. Please check your files carefully.